Ученые из KAIST (Южная корея) разработали новый подход, который значительно сокращает время расчетов сложных квантово-механических компьютерных симуляций. Благодаря новому методу, основанному на искусственном интеллекте (ИИ), ученые смогли предсказывать информацию о химических связях на атомном уровне в трехмерном пространстве.
Команда профессора Ён-Хуна Кима из Школы электротехники создала методику, основанную на нейронных сетях, которая позволяет обходить сложные алгоритмы, обычно требуемые для квантово-механических расчетов с использованием суперкомпьютеров. В традиционных расчетах плотностной функциональной теории (DFT) требуется многократное выполнение самосогласованного поля (SCF), что ограничивает применение технологии для сотен или тысяч атомов.
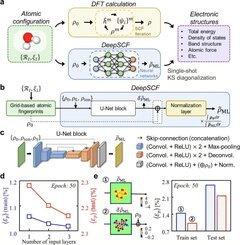
Ученые задались вопросом, возможно ли избежать SCF-процесса с помощью современных технологий искусственного интеллекта. В результате был разработан модель DeepSCF, которая обучается на информации о химических связях и ускоряет расчеты с помощью алгоритма, использующего компьютерное зрение.
Команда сосредоточилась на данных о молекулах, содержащих различные характеристики химических связей, и подготавливала их для обучения, подвергая произвольным вращениям и деформациям. В итоге они продемонстрировали эффективность и валидность новой методологии для сложных систем.